The cell-based model of coagulation is the current theory of how coagulation occurs in vivo. It is more physiological in that it explains why haemophilia is such a severe bleeding disorder, and rightly identifies factors VIII and V as co-factors for factors IX and X respectively. Detailed knowledge of the cell-based model of coagulation is not necessary for medical students or junior doctors. We illustrate it here for interest.
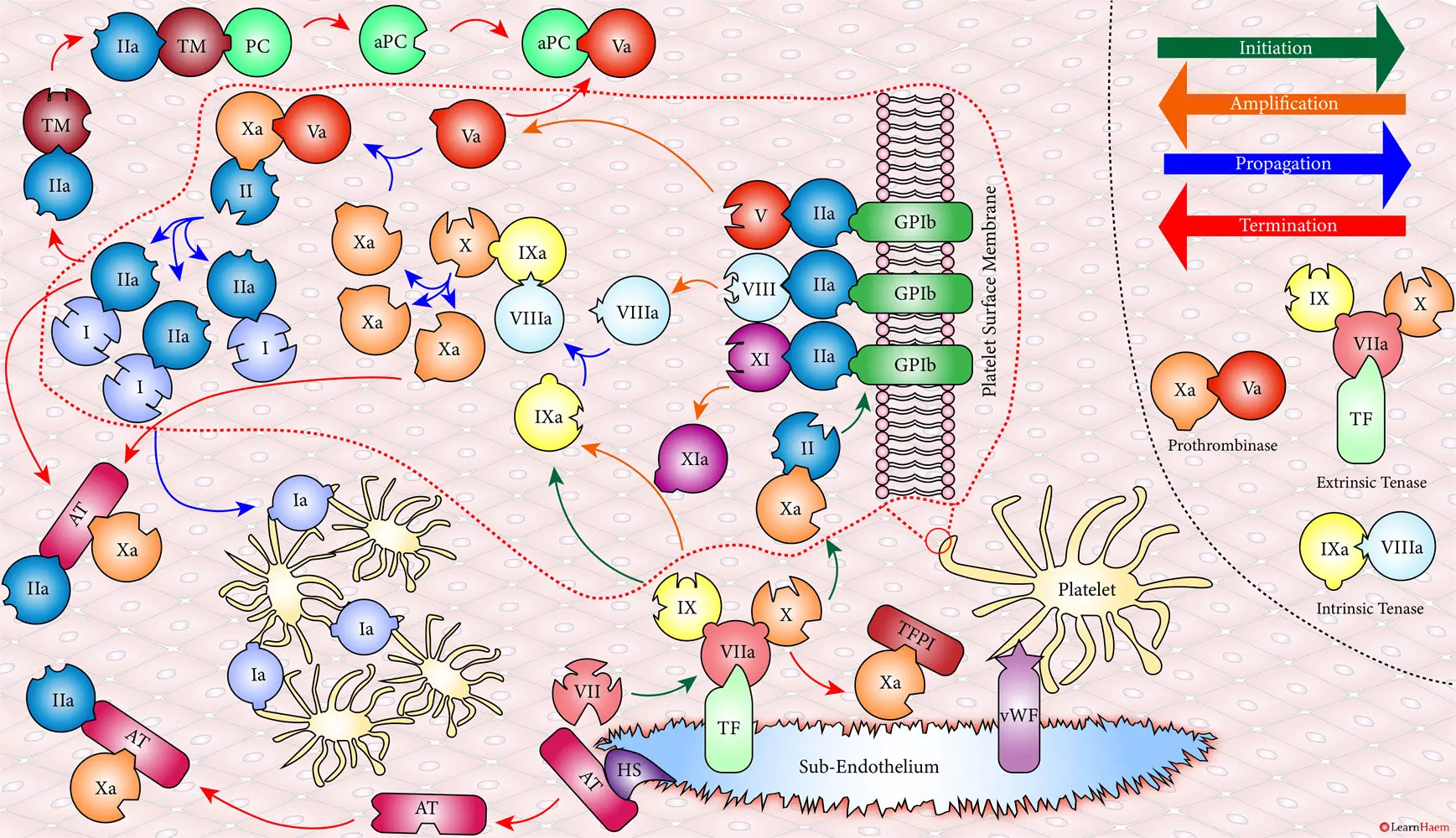
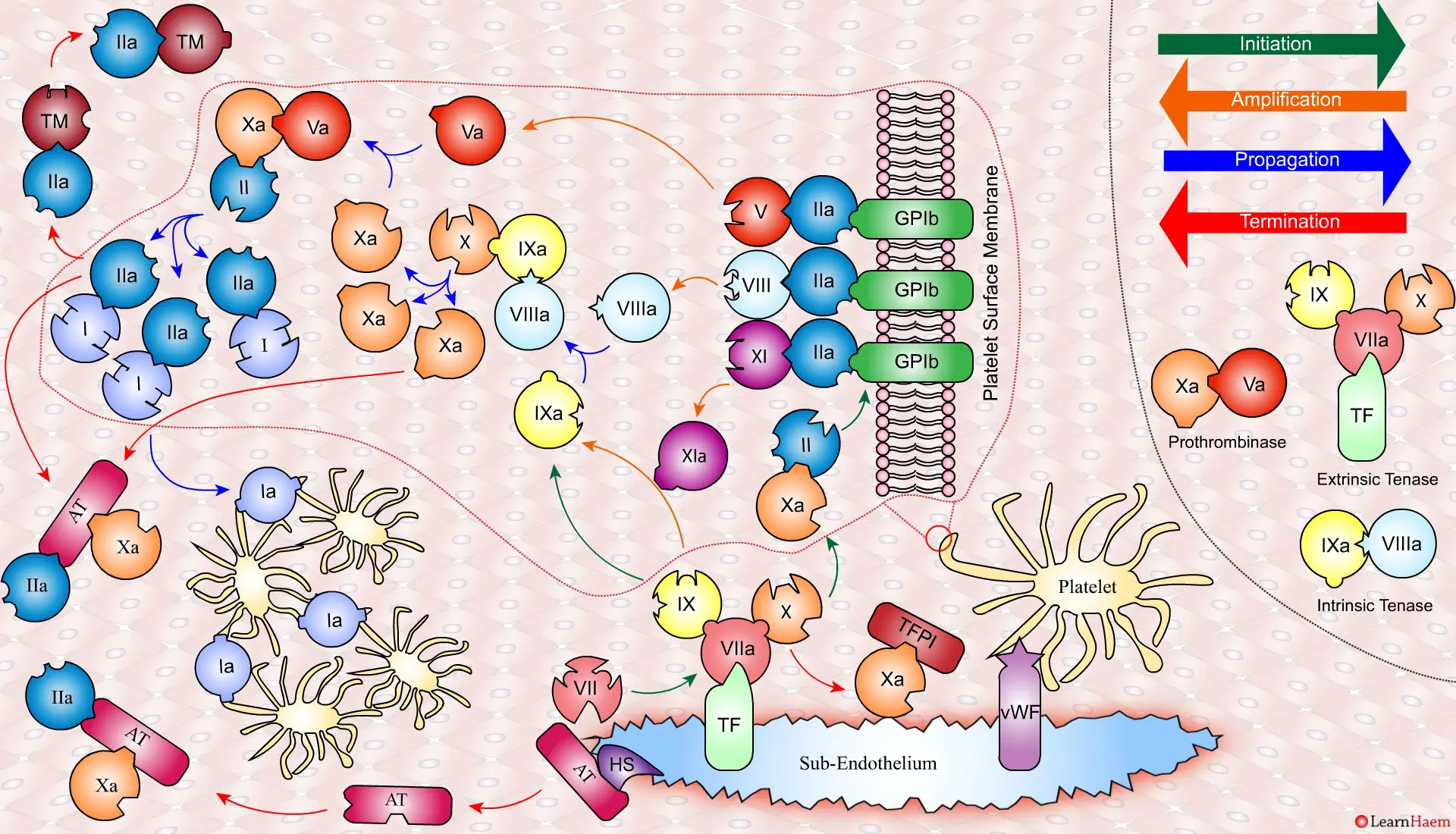
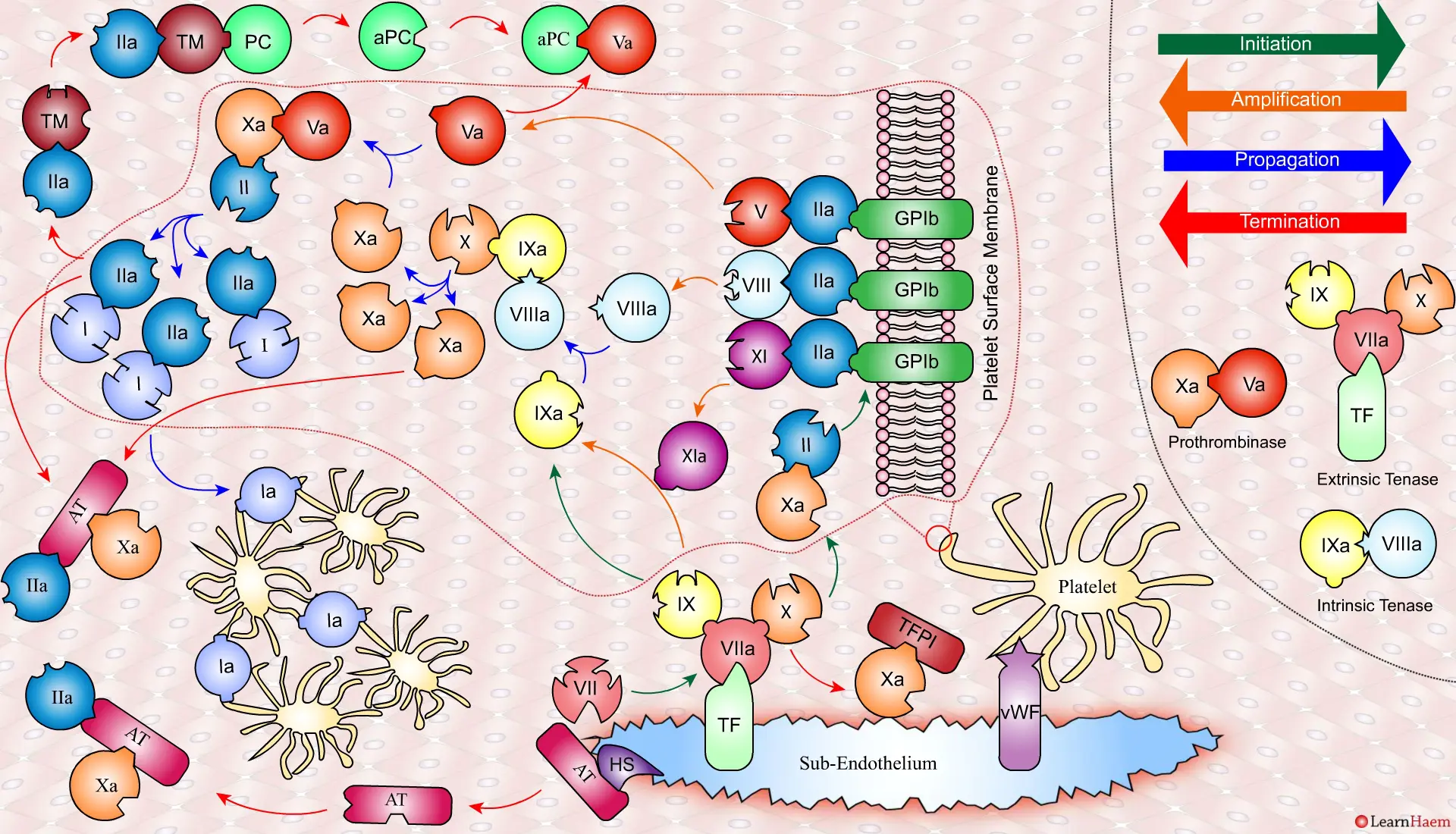
The Cell-Based Model of Coagulation.
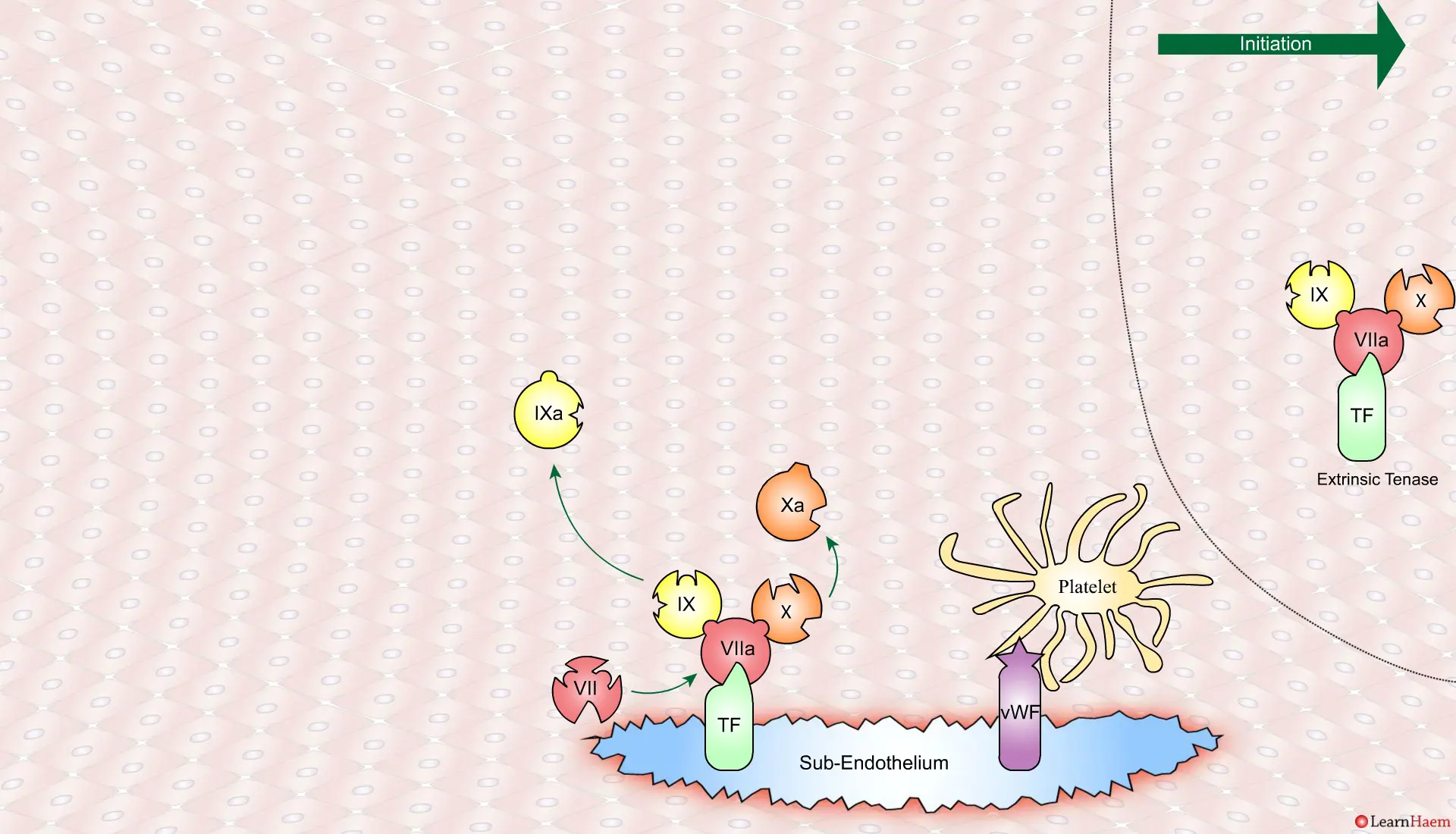
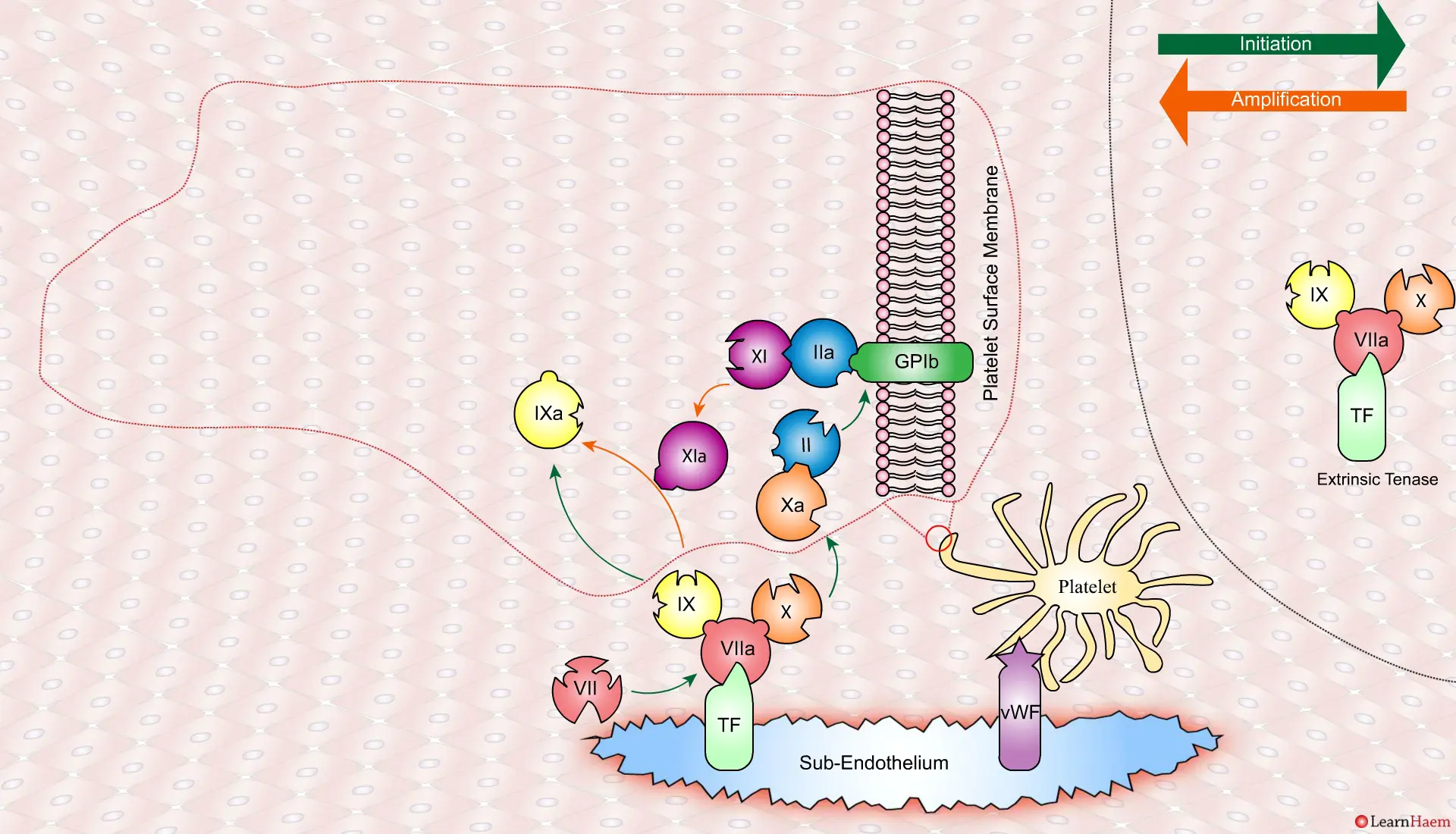
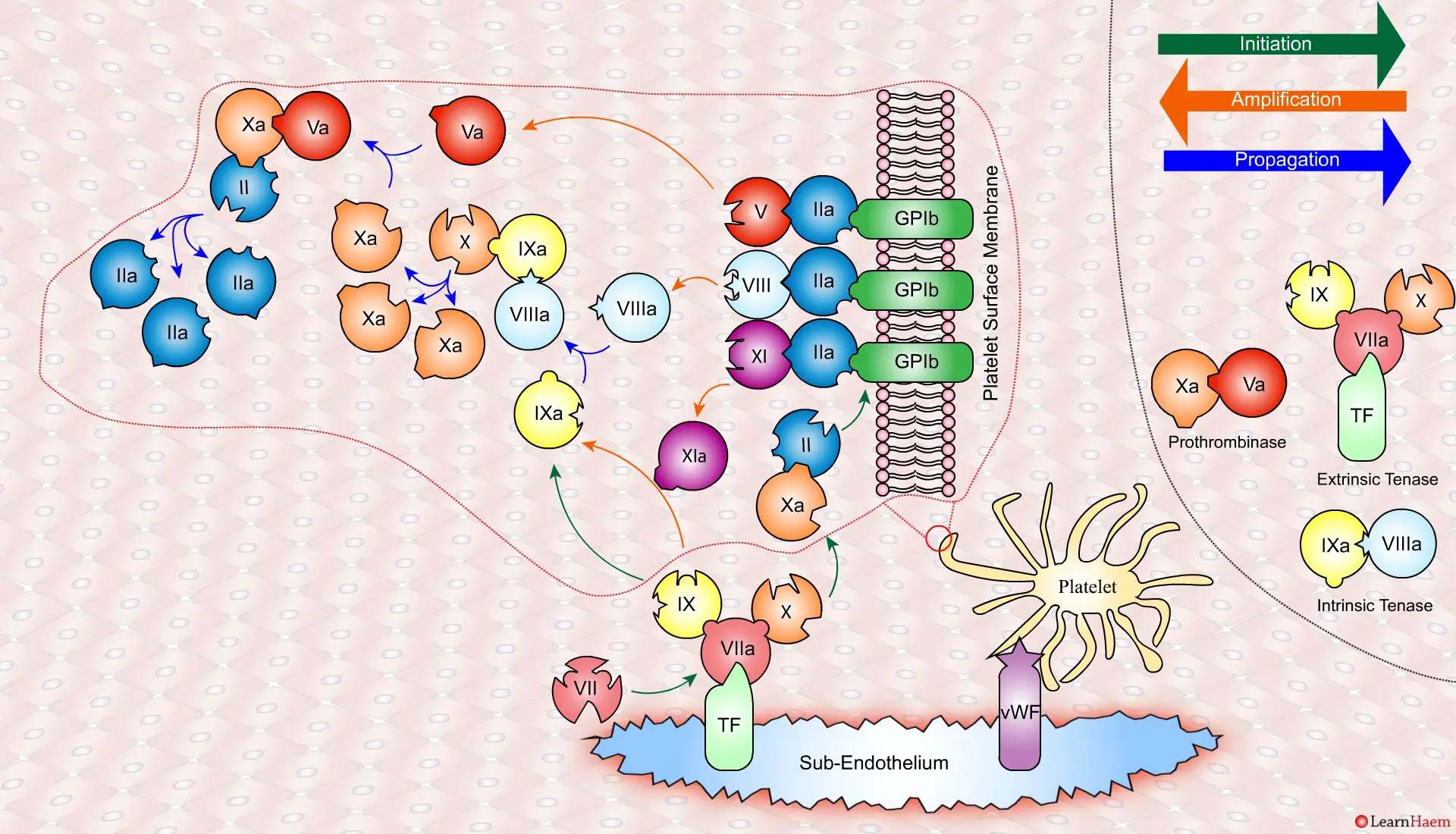
A tear in the endothelium exposes tissue factor (TF) and von Willebrand factor (vWF).
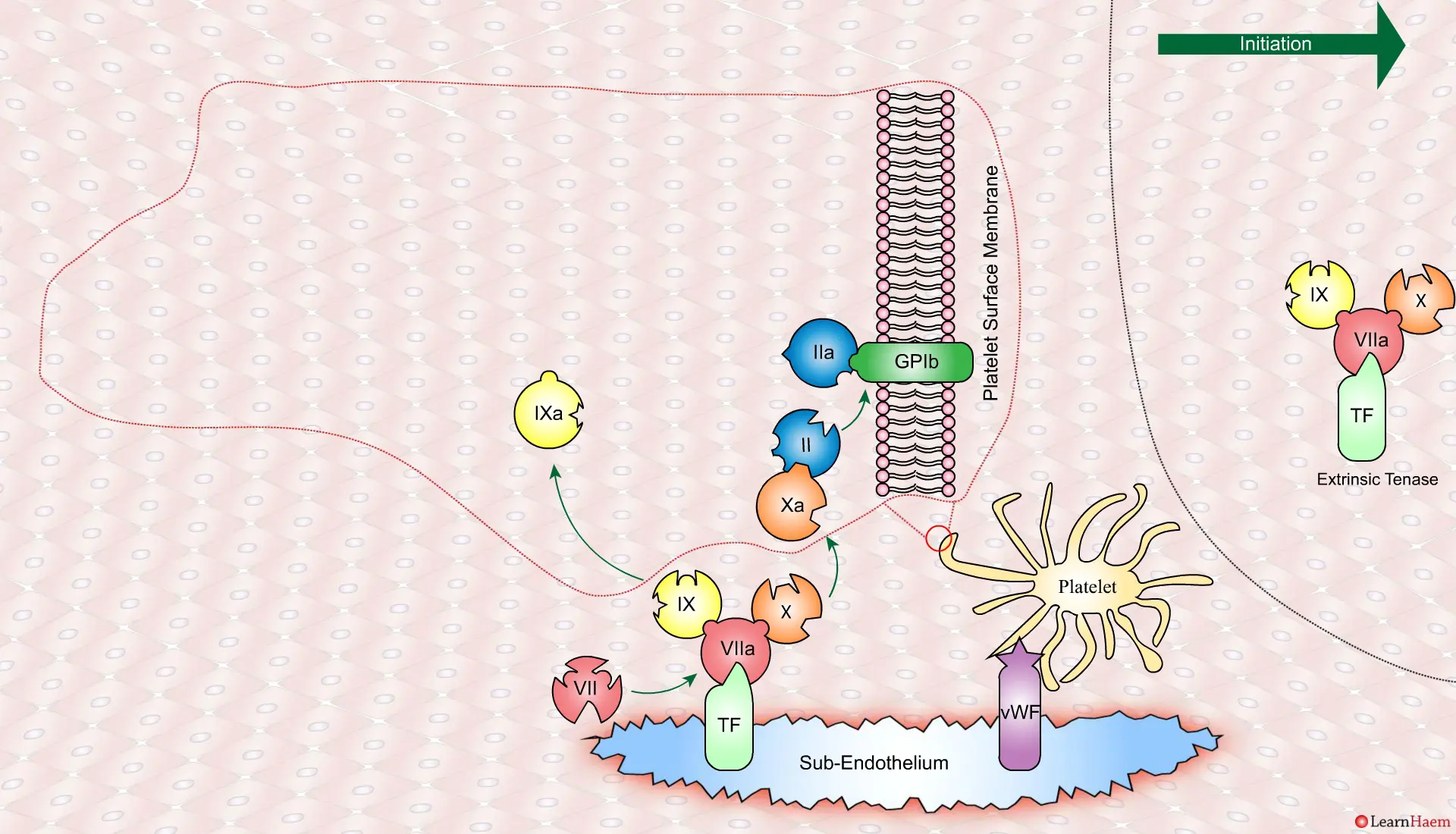
vWF binds to platelets through the GP1b receptor, anchoring them to the area of injury. The platelet membrane provides a phospholipid surface on which coagulation factors are active.
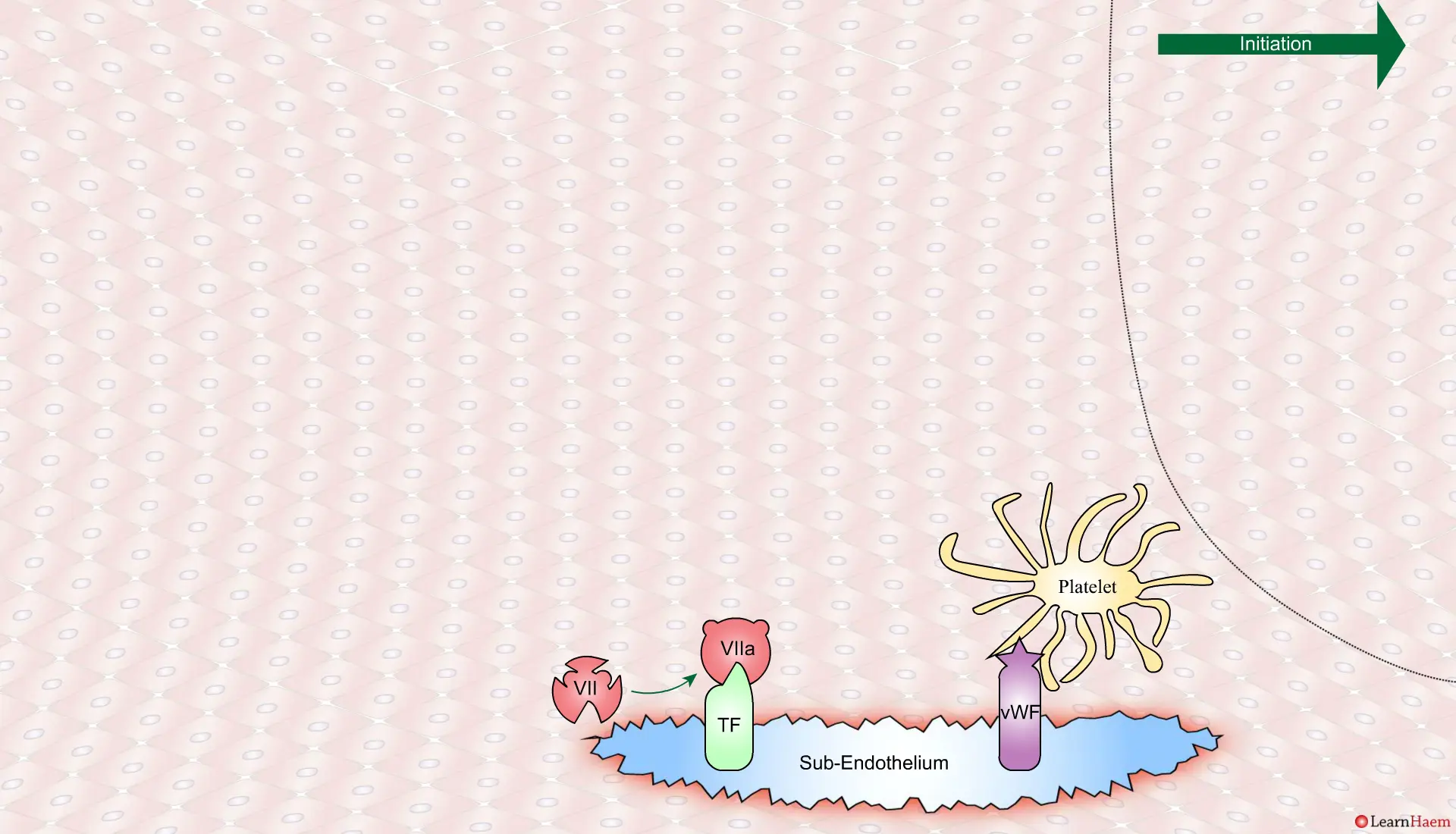
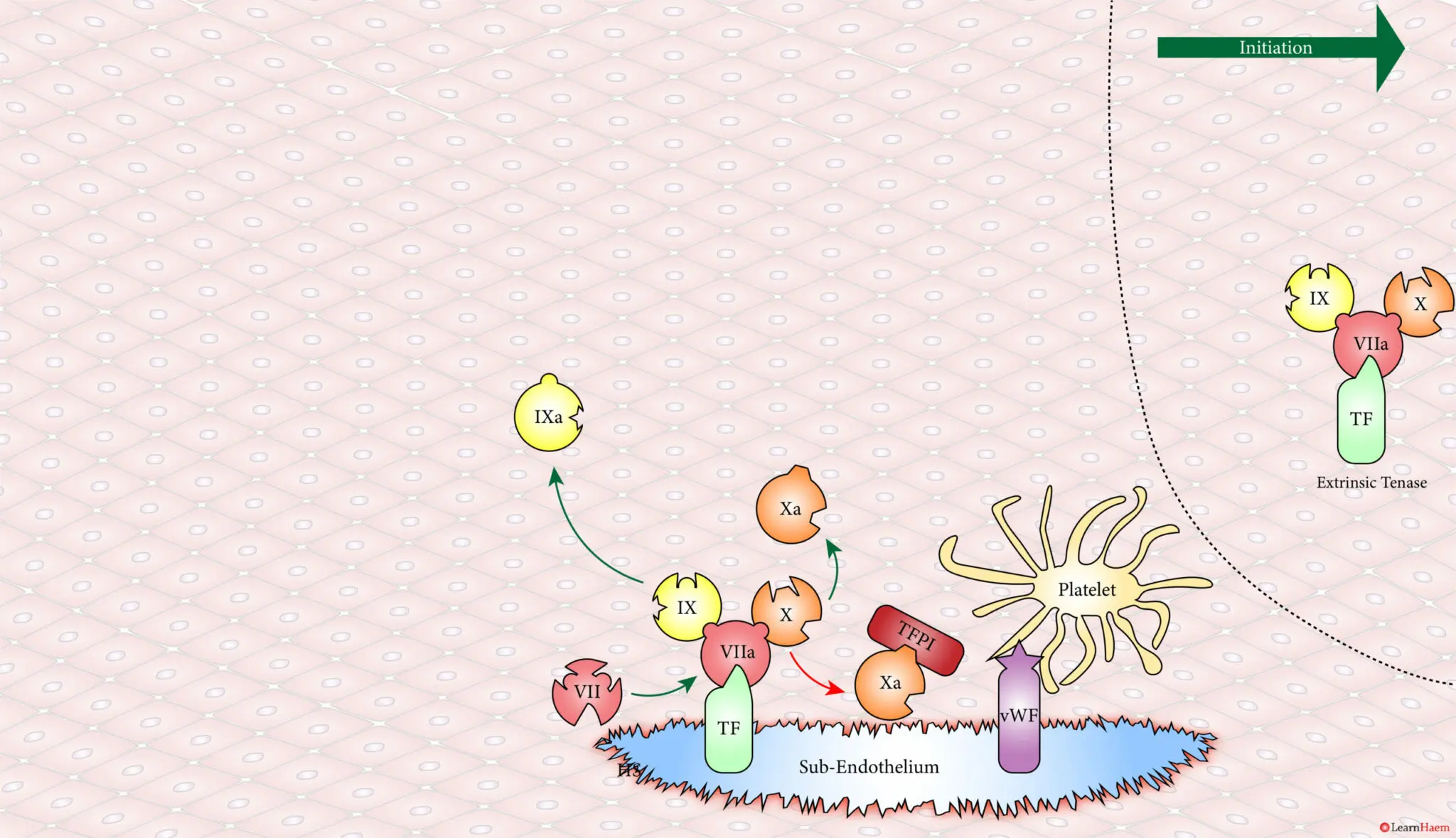
Coagulation is initiatedwhen tissue factor binds to circulating factor VII, activating it and catalysing the conversion of FIX and FX to FIXa and FXa respectively. This complex is known as the extrinsic tenase complex.
However, the FXa produced by the extrinsic tenase is very rapidly inactivated by tissue factor pathway inhibitor (TFPI). Hence, it can only catalyse the formation of small amounts of thrombin (IIa) from prothrombin (II).
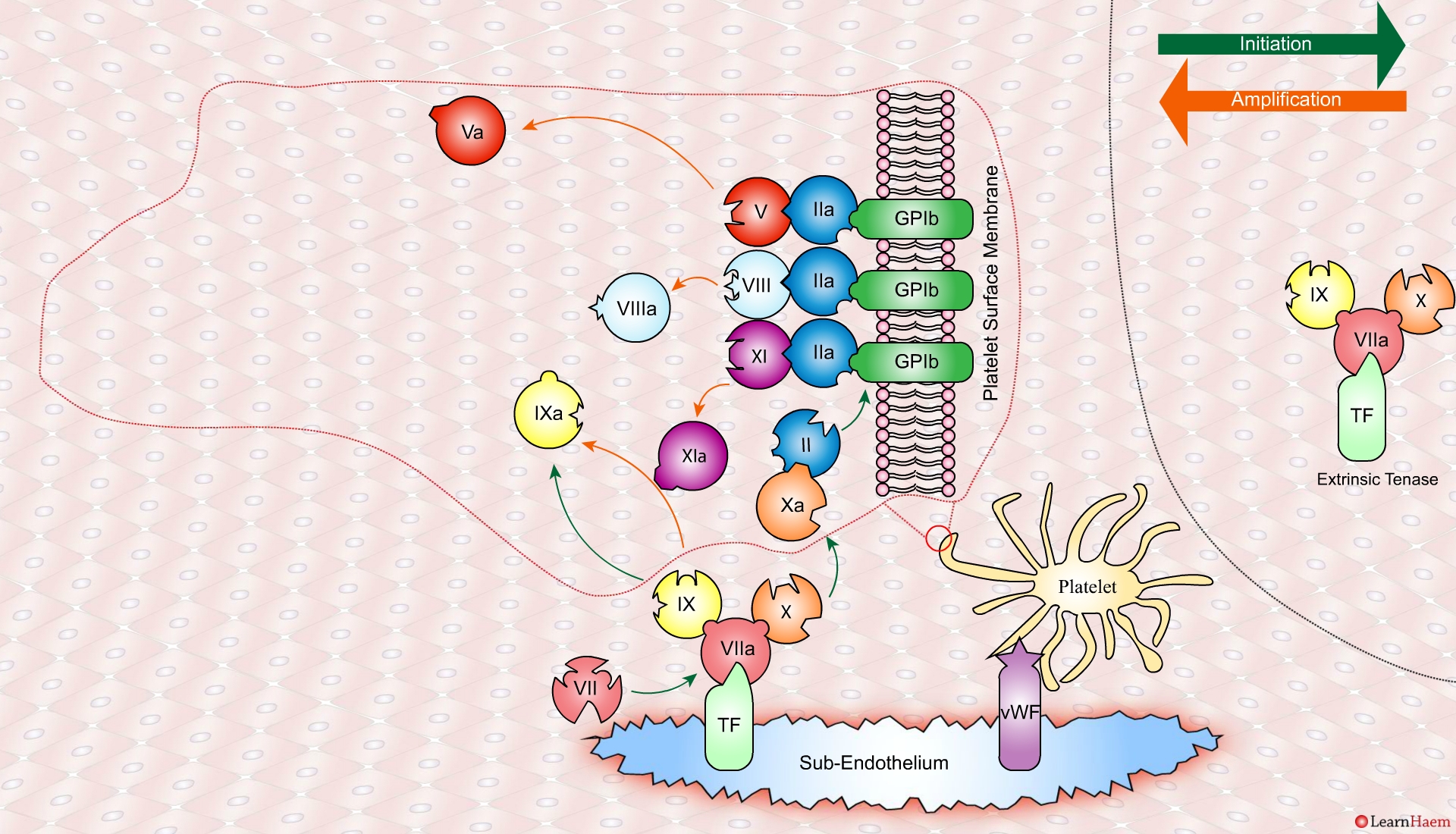
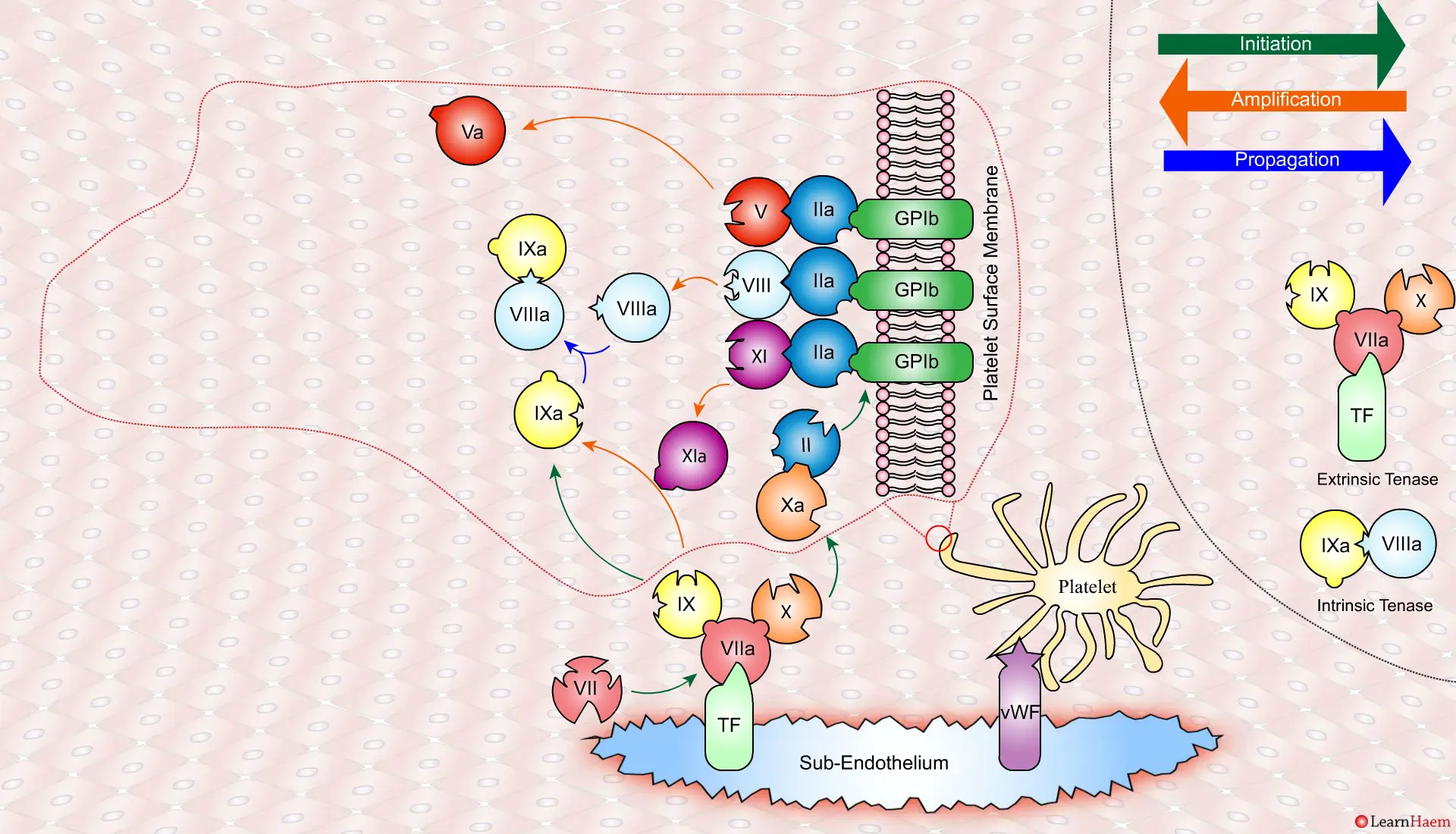
Thrombin binds to the GP1b receptors on the platelet surface, activating FXI, FVIII and FV. This results in the amplification phase.
FXIa helps to amplify the conversion of FIX to FIXa by the extrinsic tenase. The degree of amplification is variable, explaining the variable clinical phenotype of FXI deficiency.
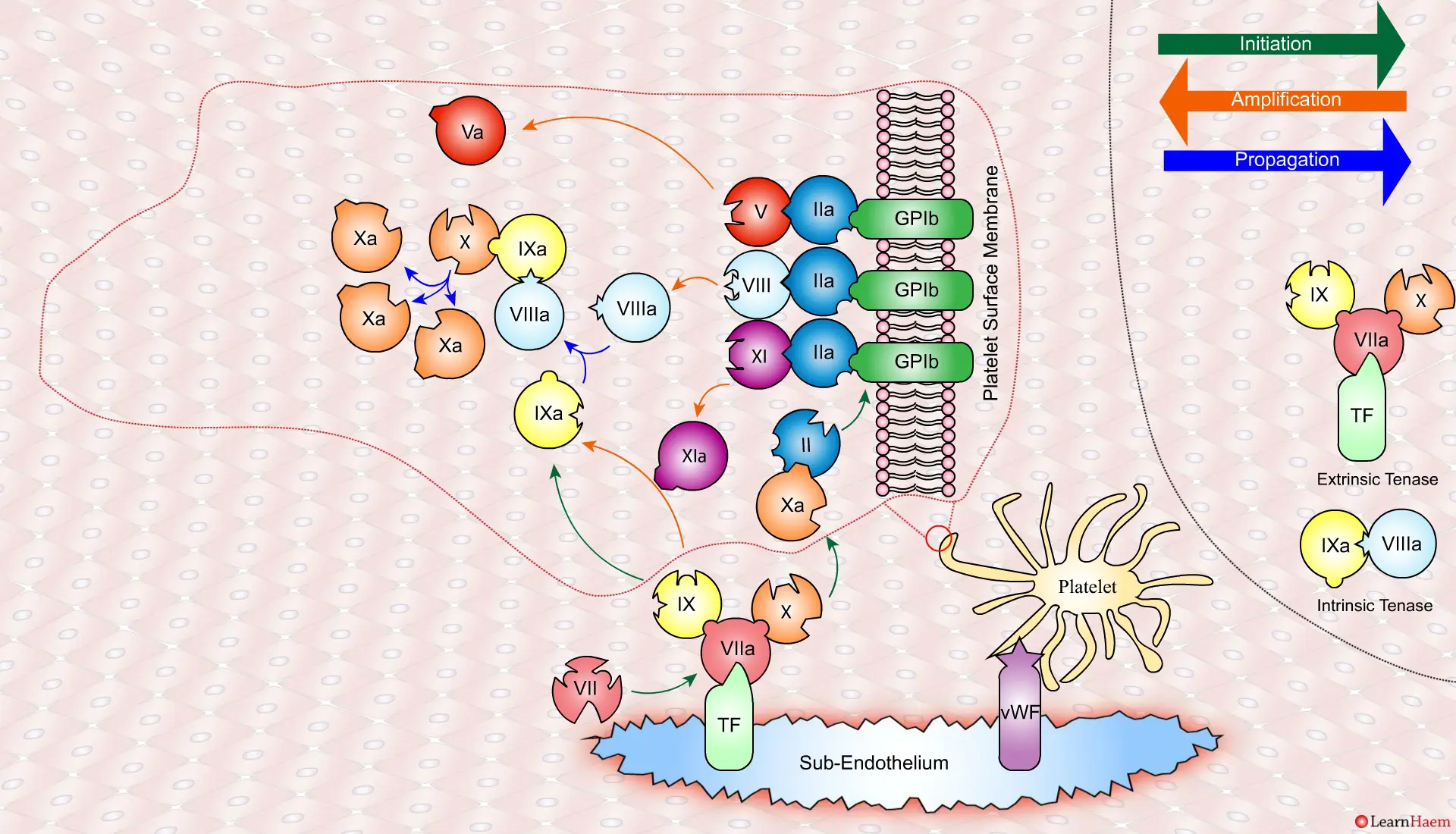
In the propagation phase, FVIIIa binds to FIXa, acting as a cofactor and resulting in the generation of the intrinisic tenase complex.
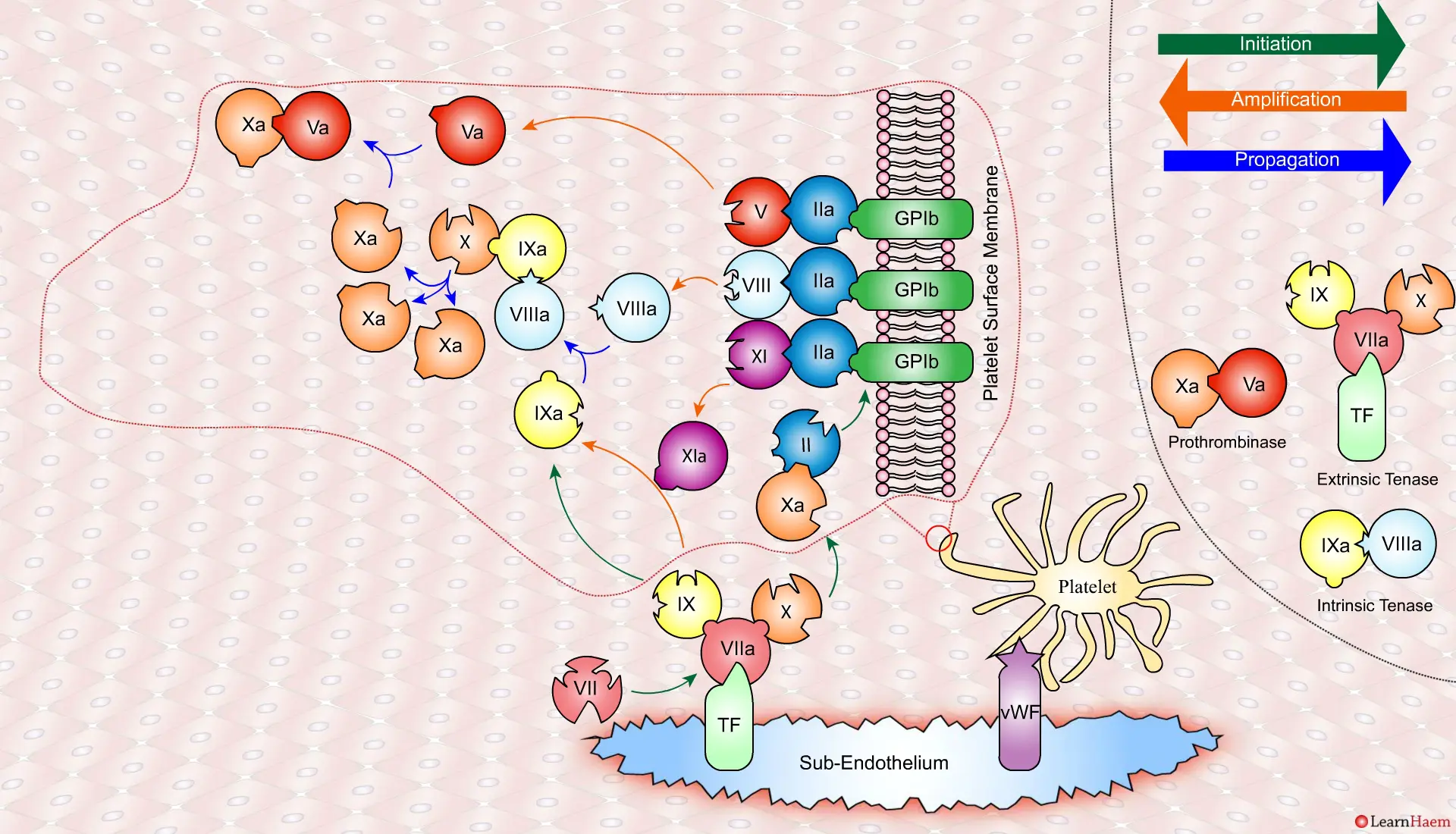
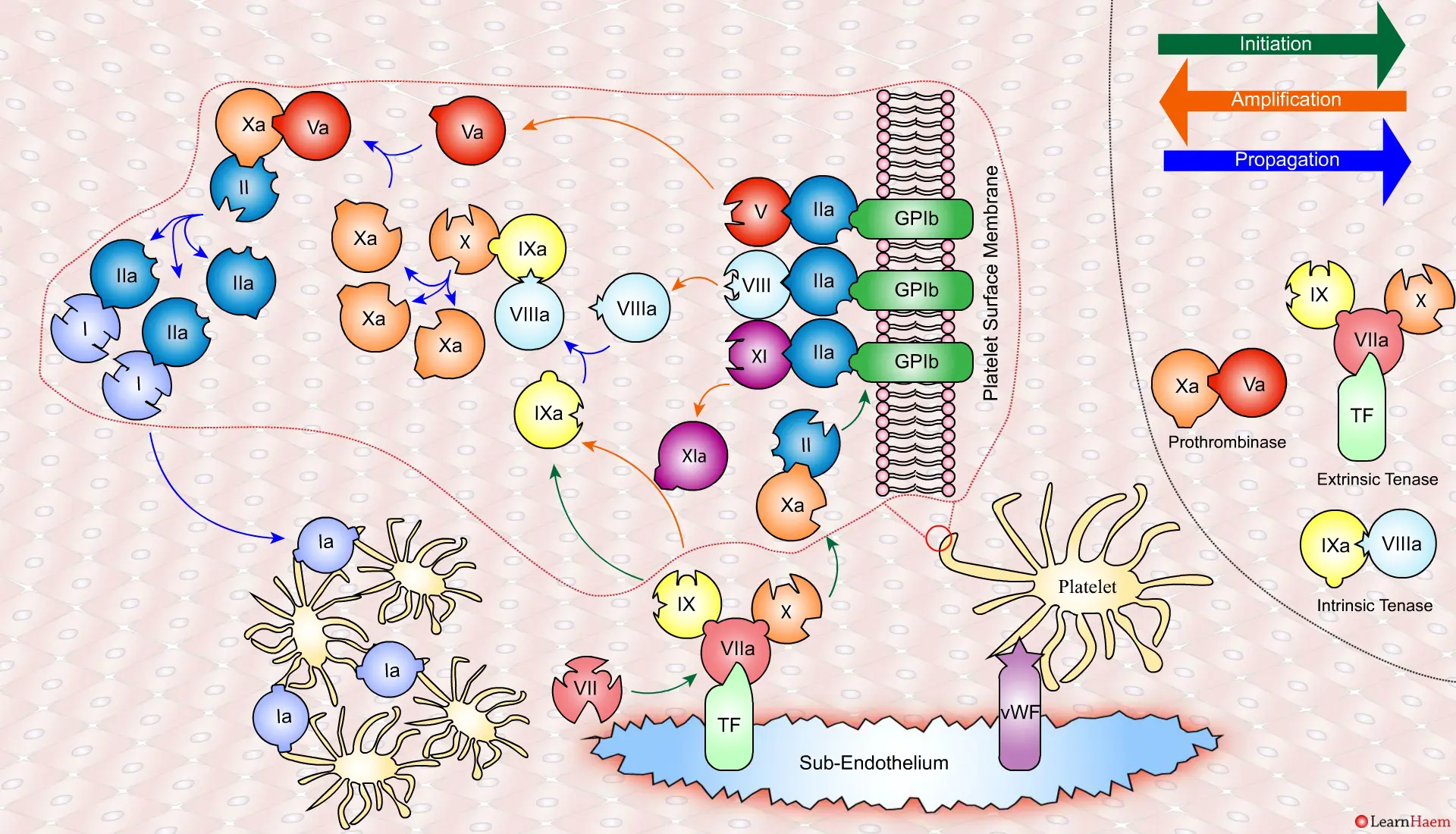
This results in the generation of large amounts of FXa from FX. The FXa combines with FVa to form the prothrombinase complex.
The prothrombinase complex catalyses the conversion of prothrombin (II) to thrombin (IIa) in large amounts.
The resultant thrombin converts fibrinogen (I) to fibrin (Ia), which cross-links platelets via the GPIIb/IIIa receptor.
This results in the formation of a platelet plug, stabilised by a fibrin mesh at the site of vessel injury.
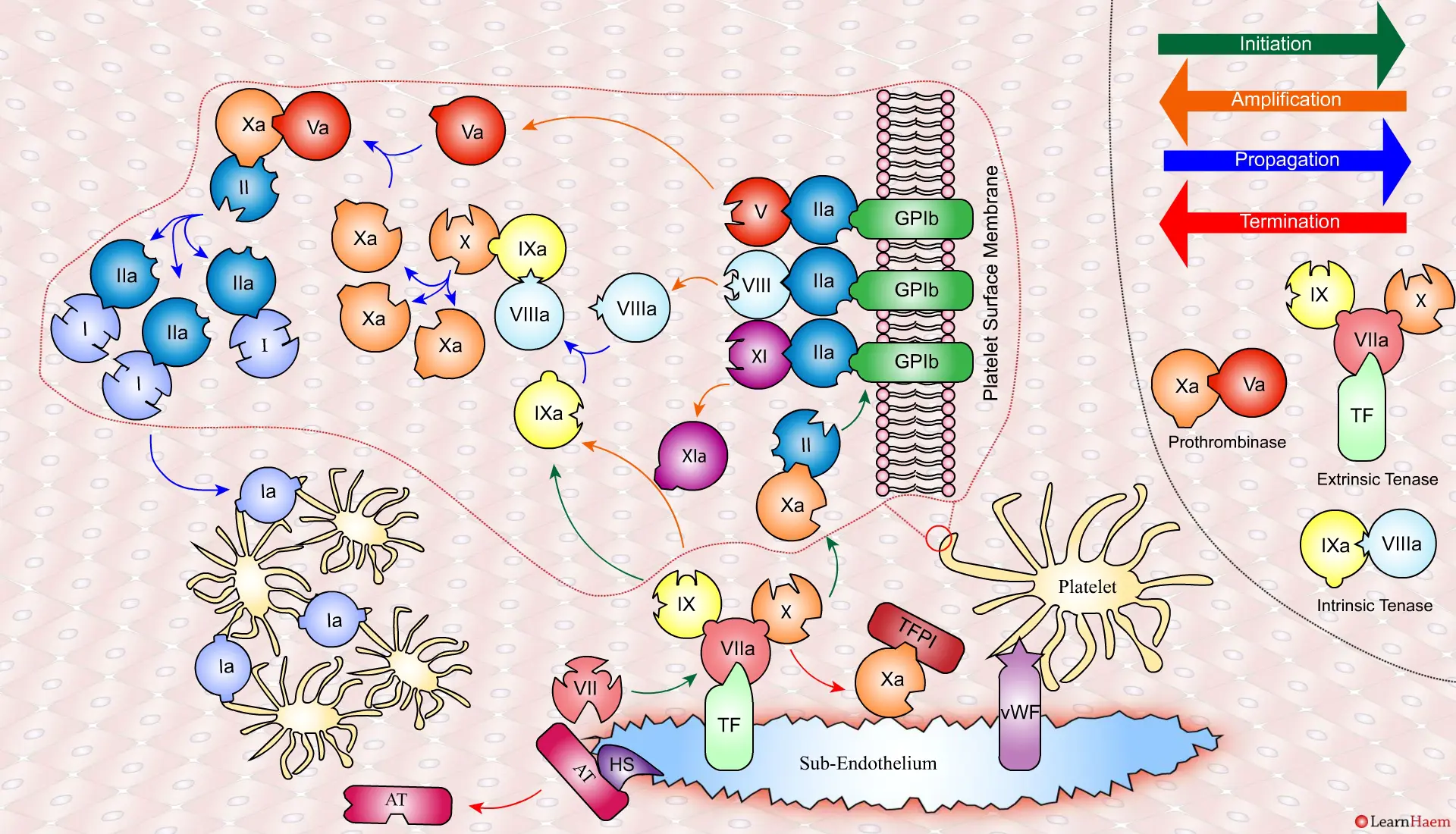
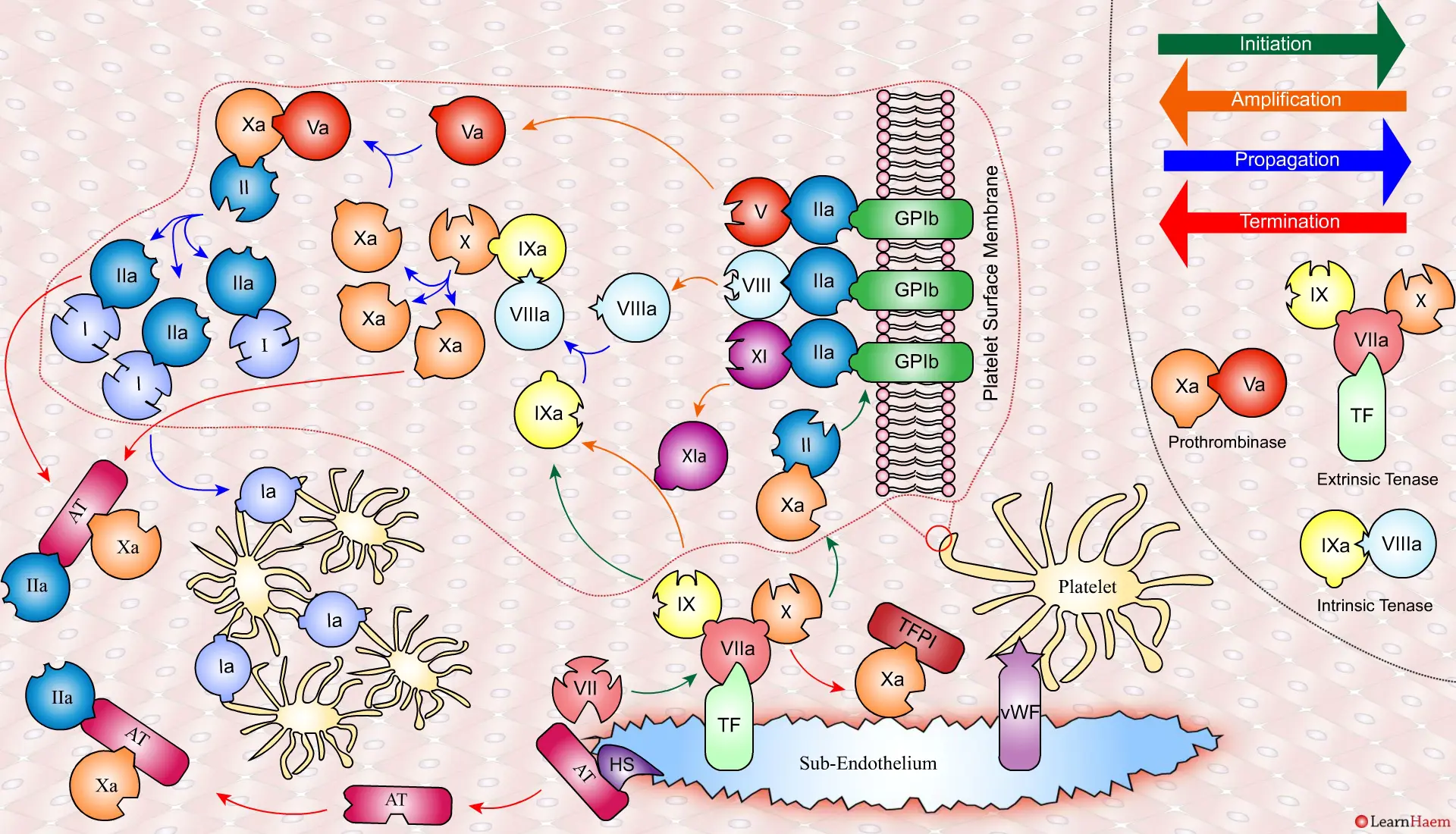
Coagulation is terminatedby two mechanisms other than TFPI.
Firstly, endothelial injury results in heparan sulphate (HS) being exposed to the blood. This induces a conformational change in circulating antithrombin (AT), which then binds thrombin (IIa) and Xa, inactivating them.
Secondly, thrombin (II) binds to thrombomodulin (TM), which is expressed on endothelial cell surfaces. This induces a conformational change in TM, which allows it to bind to protein C (PC) and activate it (aPC). aPC then binds to FVa, inactivating it. Protein S (not depicted) is a co-factor for protein C.
The video below shows an animated version of the steps described above.
FXIa helps to amplify the conversion of FIX to FIXa by the extrinsic tenase. The degree of amplification is variable, explaining the variable clinical phenotype of FIX deficiency.
I think you mean the variable phenotype of FXI deficiency rather than FIX deficiency (Haemophilia B)
Yes, thank you for highlighting this. We are currently revamping the coagulation section to make it clearer. Some of this information is targeted at Haematology trainees rather than medical students. You can view it here.


It’s the GPIIb/IIIa receptor… no IIa/IIIb
Thanks for spotting the typo!
FXIa helps to amplify the conversion of FIX to FIXa by the extrinsic tenase. The degree of amplification is variable, explaining the variable clinical phenotype of FIX deficiency.
I think you mean the variable phenotype of FXI deficiency rather than FIX deficiency (Haemophilia B)
Hello! We have reworked this page with more images to make it more user-friendly. We hope it’s useful for you!
Thank you I love the step by step. I actually understand the clotting cascade now.
Very glad you found it useful. Please let us know if there are other areas you’d like covered 🙂
Hello,
what about 1) EPCR and 2) APC inactivation of FVIIIa?
Thank you!
Yes, thank you for highlighting this. We are currently revamping the coagulation section to make it clearer. Some of this information is targeted at Haematology trainees rather than medical students. You can view it here.
Hello, I would like to use this exceptional material in my class, may I?
Yes, as long as the material remains freely accessible, for education purposes only and is credited to LearnHaem, feel free!
Hello may I know the proponents of this theory/model?
Thanks for al lthe informative content, really fantastically explained, how does calcium interact in the cell based model?
Great question – it is a key activator of coagulation, especially the tenase and prothrombinase complexes
Great presentation
Excellent speakers.
Thankyou for this wonderful summary and easy to grasp cartoons! Very helpful for exams. Will print a copy for next to the toilet