
ß Thalassaemia
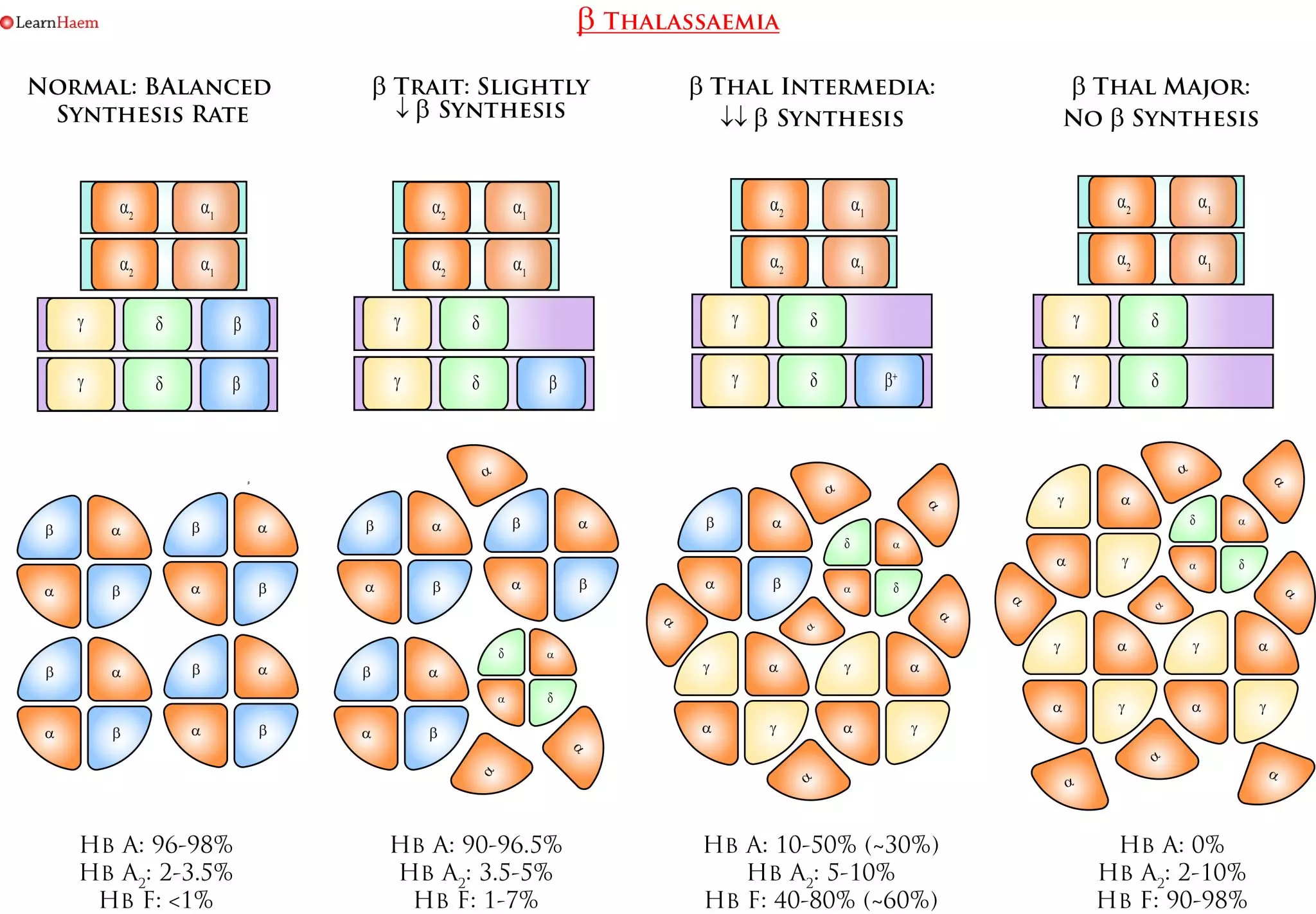
ß thalassaemia results from deletions in the ß globin gene. A normal human has 2 ß globin genes. Deletions in ß genes result in imbalanced synthesis of α globins and ß globins. As a result, excess free α globin chains accumulate and precipitate in red cell precursors, interfering with maturation and resulting in intramedullary destruction of erythroid precursors. This is known as ineffective erythropoiesis. The RBCs that enter the circulating contain varying amounts of free α globin (the amount increases with the phenotypic severity of the ß thalassaemia). The α globin and its breakdown products are deleterious to the red cell membrane, causing it to become more rigid and resulting in shortened red cell survival.
Pathophysiology

ß Thalassaemia. Deletions in the ß globin gene result in an imbalance in α and ß chain synthesis. As a result, there is excess formation of α chains. These precipitate in erythroid precursors, affecting their maturation and resulting in intramedullary destruction of erythroid precursors. Excess α chains in mature RBCs also have toxic effects on the RBC membrane, resulting in rigid cells with a shortened red cell survival.
Clinical Phenotypes

ß Thalassaemia Nomenclature. This is a simplified example of the various genotypes which can lead to the various clinical phenotypes seen in ß thalassaemia. ß thalassaemia trait occurs when one ß gene is non-functional or encodes a mutation resulting in a reduced rate of ß chain synthesis. It is characterised by a mild hypochromic, microcytic anaemia. Patients are usually asymptomatic. The red blood cell count is usually higher than expected for the degree of anaemia; this is a result of ineffective erythropoiesis. Patients with ß thalassaemia trait have an increased Hb A2 on haemoglobin electrophoresis. However, care must be taken in interpreting the results when there is a concurrent iron deficiency, as iron deficiency lowers the Hb A2 fraction. The difference between ß thalassaemia intermedia and major is transfusion-dependence. Patients with ß thalassaemia intermedia (non-transfusion-dependent thalassaemia, NTDT), have a moderate anaemia with hepatosplenomegaly. There is ongoing haemolysis, and they may have features of extra-medullary haematopoiesis. They can become iron overloaded. Patients with ß thalassaemia major are transfusion dependent because of the severe or absent ß chain production. They have a characteristic facies which results from expansion of the bone marrow in childhood. They have a severe anaemia and die in childhood if a transfusion programme is not initiated. They are susceptible to iron overload and its consequences.
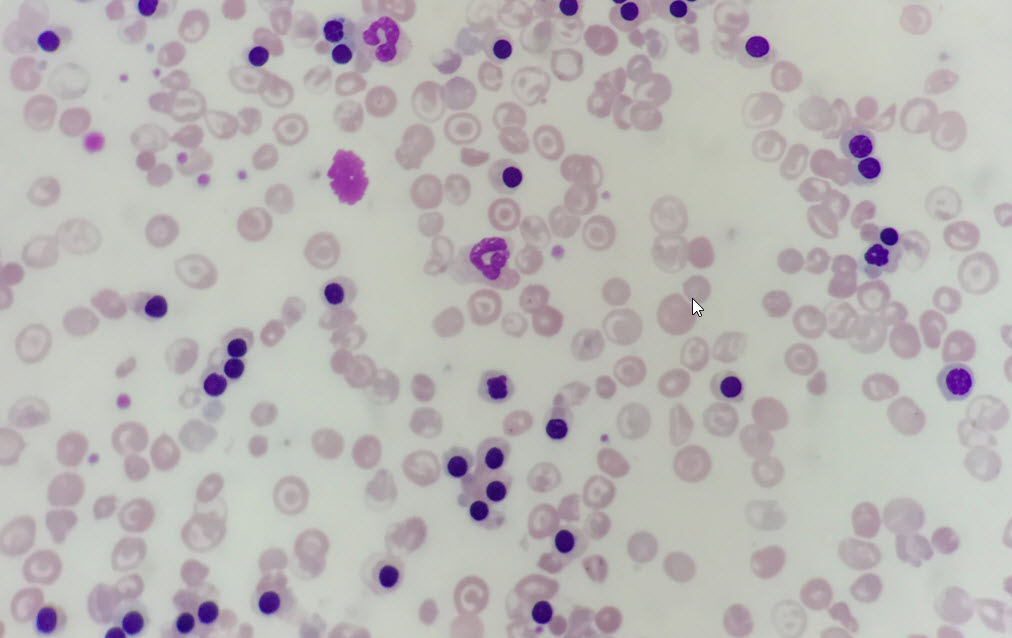
Peripheral Blood Film of a Patient with ß Thalassaemia Intermedia. There are numerous circulating nucleated red blood cells; the patient was actively haemolysing due to a concurrent chest infection. There are numerous target cells in the background. These are seen in haemoglobinopathies and are two-dimensional representations of a bell-shaped RBC that exists in vivo. There is also significant anisopoikilocytosis (cells of different shapes and sizes) and polychromasia.
Inheritance
ß thalassaemia is inherited in an autosomal recessive fashion. Because there are a number of mutations in the ß globin gene that can result in ß thalassaemia, the exact clinical phenotype depends on whether the ß globin gene is functional, and the severity of the functional impairment. The example below shows a patient with ß thalassaemia intermedia from ß+/ß0 and her husband with ß thalassaemia trait.
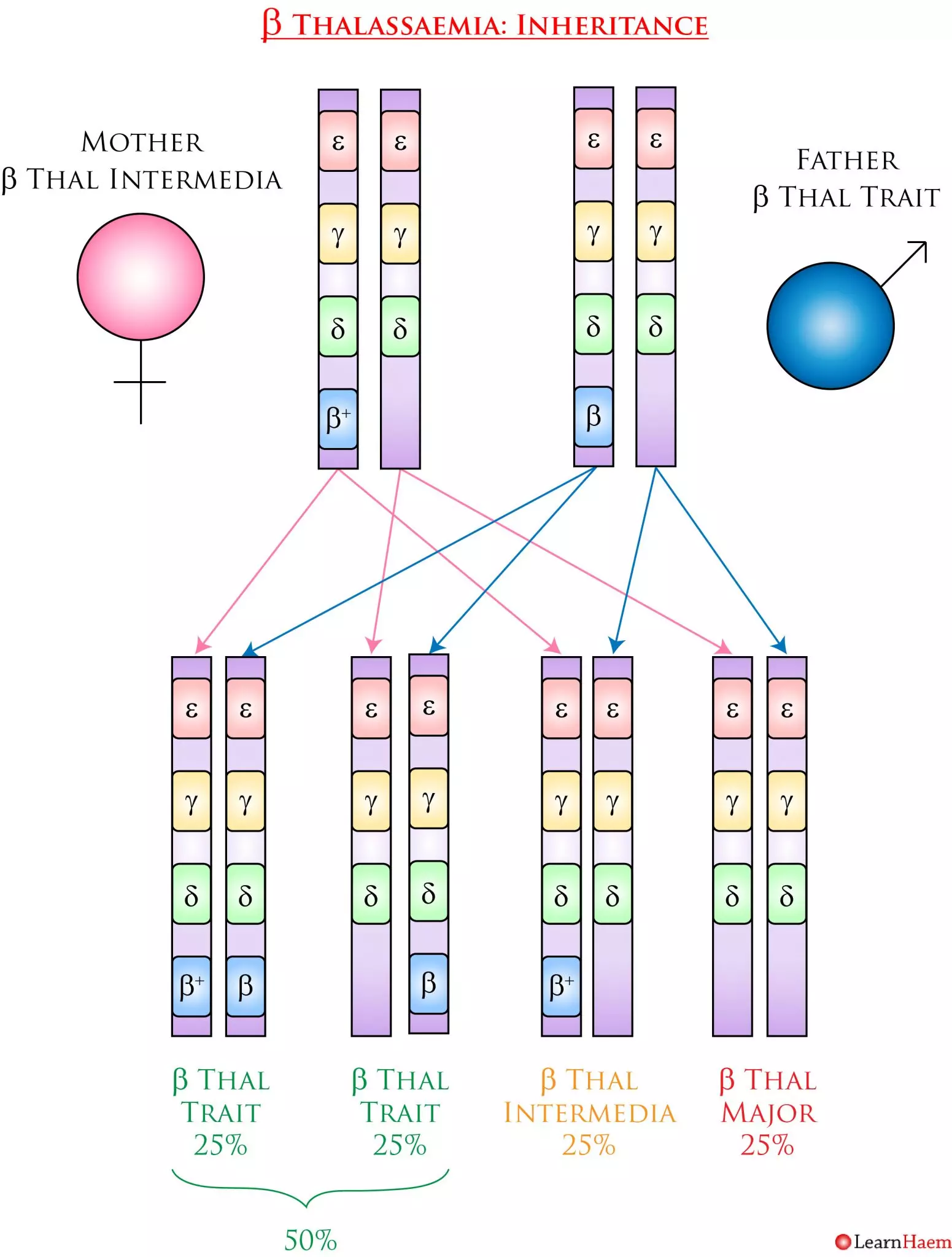
ß Thalassaemia Inheritance Pattern. In this example, the mother has ß thalassaemia intermedia (ß+/ß0). Her partner has ß thalassaemia trait (ß/ß0). They have a 25% chance of having a child with ß thalassaemia major, a 25% chance of having a child with ß thalassaemia intermedia, and a 50% chance of having a child with ß thalassaemia trait.
The letter ß (Eszett letter) is used only in German, and can be typographically replaced with the double-s digraph ⟨ss⟩ and is not the same as β representing beta.
Thanks for the otherwise clear and concise explanation of the thalassemias