
Sickle Cell Anaemia
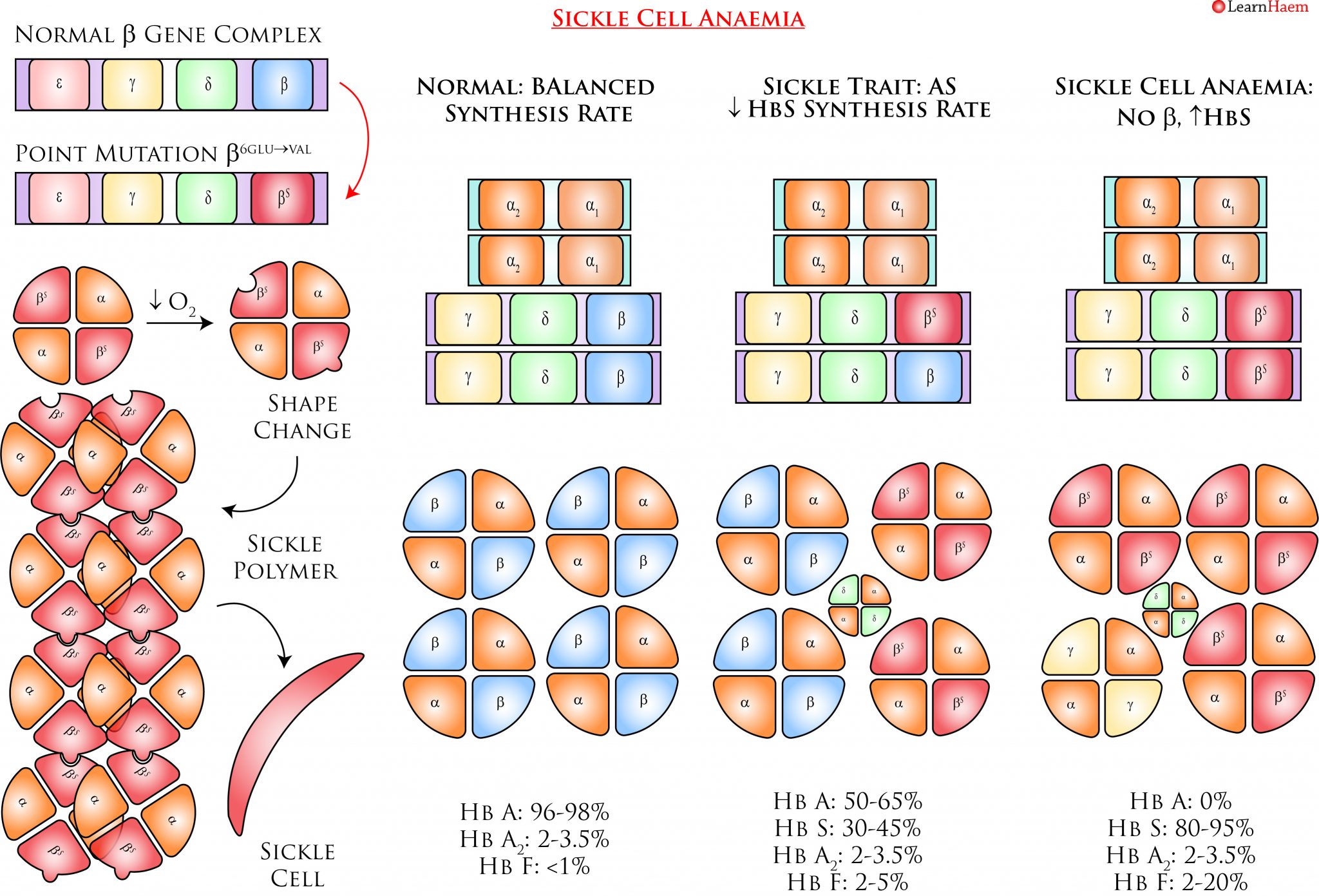
Sickle cell anaemia results from a point mutation at position 6 of the ß globin gene (β6glu→val). It is inherited in an autosomal recessive manner. Heterozygotes for this mutation have sickle cell trait, which is largely asymptomatic. Homozygotes have sickle cell anaemia, which is characterised by sickling and vaso-occlusion.
Pathophysiology
The β6glu→val mutation results in a variant ß globin known as βS. This interacts with α globin to form HbS (αα/βSβS). HbS is susceptible to deoxygenation, which triggers a conformational change that forms the basis for the pathogenic consequences of sickle cell anaemia.

Sickle Cell Anaemia. Sickle cell anaemia is caused by a point mutation on the ß globin gene. The resultant haemoglobin is both a variant haemoglobin and one which is synthesized at a slower rate, which explains why the fraction of HbS in individuals with sickle cell trait is only ~30-45%. HbS is susceptible to deoxygenation, which triggers a conformational change in the protein. This conformational change renders the haemoglobin susceptible to polymerisation, as there are complementary binding sites on other HbS molecules which are also unmasked by deoxygenation. As HbS polymerises, the red cell elongates and stiffens into a sickle shape. This makes red cells more rigid and less deformable, thus resulting in vaso-occlusion when these cells adhere and become trapped to the vascular endothelium.
Leave A Comment