
Alpha thalassaemia results from deletions in the α globin gene. It is frequently found in Southeast Asian and Mediterranean populations. A normal human has four α globin genes. Deletions in α genes result in imbalanced synthesis of α globins and ß globins. In more severe disease (–/-α or –/–), the excess ß globins for HbH (ß4) and Hb Barts (γ4) respectively. These haemoglobins have an extremely high oxygen affinity and are unstable, precipitating in red blood cells as they age. This results in membrane damage and markedly reduced red cell survival. The high oxygen affinity of HbH and Hb Barts means they are ineffective as oxygen delivery devices.
Single (αα/-α, heterozygous α+ thalassaemia) or two (αα/–, α0 thalassaemia, or -α/-α, homozygous α+ thalassaemia) gene deletions result in α thalassaemia trait. This is characterised by microcytosis, and occasional target cells. In single gene deletions, the Hb is normal. Two gene deletions may result in a mild anaemia.


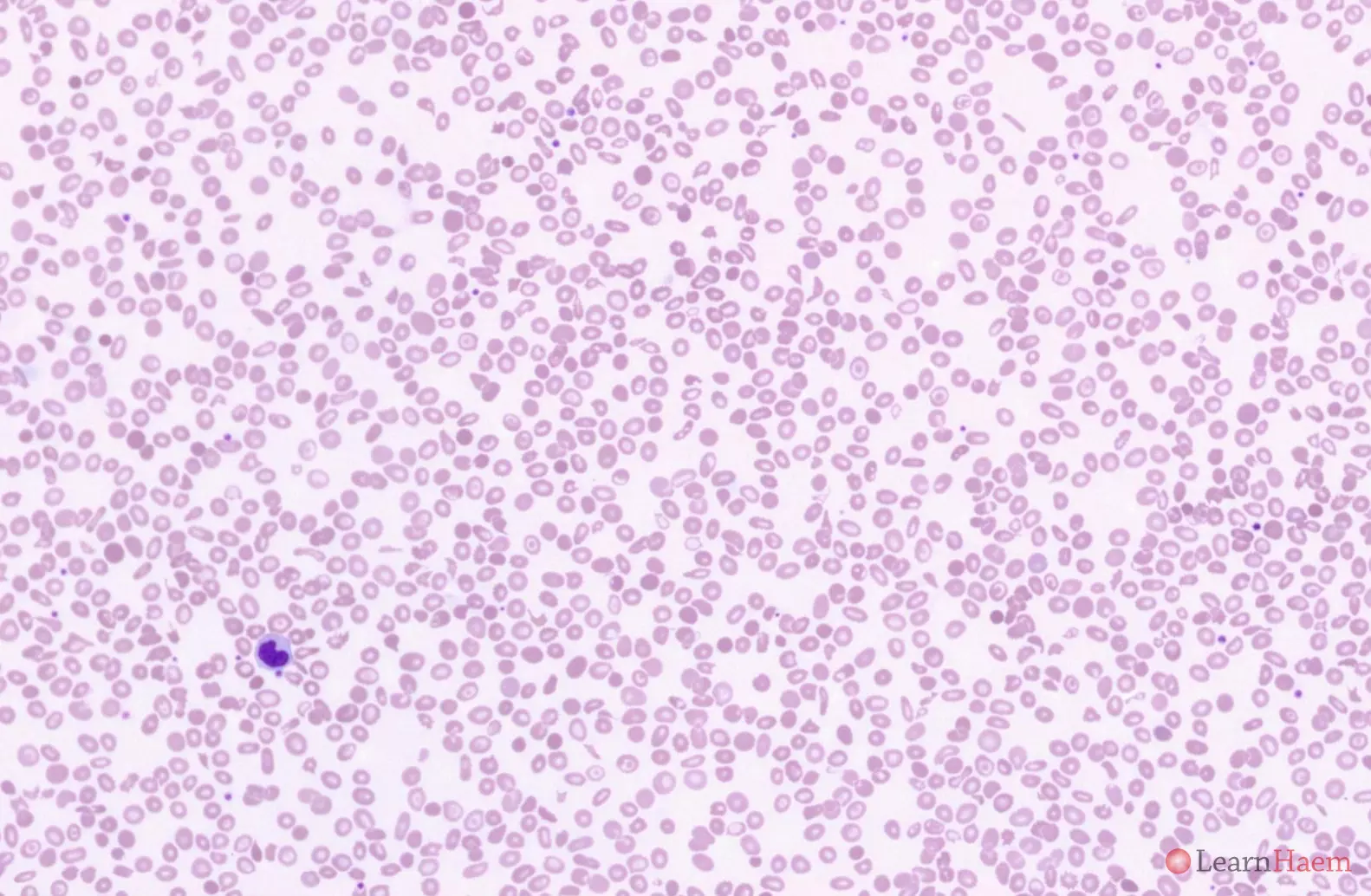
This is the blood film from a patient with Haemoglobin H disease. HbH disease is characterised by a moderate anaemia (Hb typically ~80g/L, but can range from 60-100g/L). Haemolytic episodes are triggered by infections, especially parvovirus B19 infection and exposure to oxidant drugs. Pregnancy exacerbates the anaemia due to a dilutional effect.
Blood film features:
- Marked hypochromic, microcytic anaemia
- Anisopoikilocytosis with target cells, teardrop cells and fragments
- Basophilic stippling
- Polychromasia
Differential diagnosis:
- Iron deficiency anaemia (degree of anisopoikilocytosis is too severe for isolated iron deficiency).
- ß thalassaemia intermedia (such patients tend to have circulating nRBCs, which are absent in HbH disease. The reticulocytosis in HbH disease is also more prominent.).
- Hereditary pyropoikilocytosis (cells are normocytic and haemoglobinisation is normal).
- Congenital dyserythropoietic anaemias (often characterised by macrocytosis with no reticulocytosis).
- Acquired HbH disease (look for features of dysplasia, which may point towards an underlying myelodysplastic syndrome).
Haemoglobin electrophoresis:

Alkaline gel electrophoresis from a patient with haemoglobin H disease showing a fast band corresponding to haemoglobin H. The Hb A2 band is also very faint, a typical feature of HbH disease.

Acid gel electrophoresis from a patient with HbH disease, showing no abnormal bands. HbH has the same mobility as Hb A.
HPLC:
HPLC from a patient with HbH disease, showing a variant haemoglobin (haemoglobin H, red arrow) with a very short retention time.
Further tests:
- HbH inclusions can be observed in HbH disease and in some cases of α thalassaemia trait. These are seen using supravital stains such as brilliant cresyl blue or new methylene blue, which highlight ribosomal precipitates in red blood cells.
- Haemoglobin electrophoresis is normal in deletional α thalassaemia trait.
- Hb Constant Spring is a mutational α thalassaemia which gives rise to a thalassaemia trait phenotype. It gives rise to a slow band on haemoglobin electrophoresis.
- In HbH disease, a fast band corresponding to haemoglobin H can be observed.
- HPLC can also identify HbH and Hb Constant Spring.
- Confirmation of α thalassaemia is best done via genetic testing for high-risk patients.
Other resources:
- PHE Family Origin Questionnaire
- PHE Sickle Cell and Thalassaemia Screening Programme (2019)
- BCSH Guideline: Significant Haemoglobinopathies: Guidelines for Screening and Diagnosis (2010)
- BCSH Guideline: Red blood cell specifications for patients with hemoglobinopathies: a systematic review and guideline (2020)
- TIF Guideline: Management of Non-Transfusion Dependent Thalassaemia (2017)
- ASH Image Bank: HbH Disease
Leave A Comment